Genetic Analysis
What is Genetic Analysis?
Genetic analysis is a term used to describe the study of a sample of DNA to look at differences, or variants, that may increase an individual's risk for disease or impact drug responses.
While some genetic diseases, like Huntington's disease, are due to single gene changes, most inherited diseases arise from a number of genetic changes that can be compounded by environmental factors, making it difficult for researchers to track down all the related factors.
Although any two people are 99.9% identical at the genetic level, the 0.1 percent difference may help explain why one person is more susceptible to a specific disease than another person. Studying patterns of genetic variation can help scientists determine which differences are disease-related.
Introduction to NGS
With its unprecedented throughput, scalability, and speed, next-generation sequencing (NGS) enables biological studies at a level never before possible.
Learn MoreUnderstanding Genetic Variation and Disease Biology
Extensive resources have been invested in sequencing the human genome and various model organisms. Further resources are now being directed towards increasing our understanding of genetic variation and function, and using these findings to develop future new diagnostic methods and therapeutics.
There's a continuing need to analyze massive numbers of genetic markers and patient samples, in an effort to understand the underpinnings of major diseases. Illumina technologies are uniquely positioned for use in these studies, and are helping further our understanding of genetic variation at the DNA, RNA, and protein levels.
Illumina Microarray Technology
BeadArray technology enables high-density BeadChip arrays to facilitate diverse applications.
Learn MoreDNA, RNA, and Protein Basics
The human body is composed of billions of cells, each containing deoxyribonucleic acid (DNA) which encodes the basic instructions for cellular function. The complete set of an organism's DNA is called its genome.
The human genome is organized into 23 pairs of chromosomes which are further divided into over 30,000 smaller regions called genes. Each gene is comprised of a string of nucleotide bases labeled A, C, G and T. Human DNA has approximately 3 billion nucleotide bases and their precise order is known as the DNA sequence.
When a gene is "expressed," a partial copy of its DNA sequence—called messenger RNA (ribonucleic acid) or mRNA—is used as a template to direct the synthesis of a protein.
Proteins, in turn, direct all cellular function.
Complex Disease Research
Find array and NGS solutions to advance your complex genetic disease research in autoimmune and rheumatic diseases, heart disease, neurological disorders, psychiatric disorders, and more on a molecular level.
Learn MoreWhat is a SNP?
Single nucleotide polymorphisms (SNPs) are single nucleotide (A, G, C, or T) changes in DNA. SNPs are by far the most common source of genetic variation. The human genome is thought to contain over 10 million SNPs, about one in every 300 bases.
Many genetic analysis tools are available to interrogate SNPs. Researchers can analyze hundreds of thousands of SNPs across the entire genome and compare patterns between healthy and diseased patient samples. Follow-on fine-mapping studies may involve analysis of a smaller number of SNPs in defined regions of the genome, in an attempt to find causative SNPs for a given disease.
What is Gene Expression?
Gene expression analysis is the process of determining which genes are active in a specific cell or group of cells. It is commonly studied by measuring messenger ribonucleic acid (mRNA), the intermediary between genes and proteins.
By comparing gene expression patterns between cells from different environments, such as normal tissue vs. diseased tissue, researchers can determine which genes are active or inactive in various disease states.
What is DNA Methylation?
DNA methylation is the attachment of methyl groups to cytosine nucleotides at CpG sites in the genome. DNA methylation is thought to affect gene transcriptional regulation and can be heritable.
Since faulty DNA methylation is associated with a variety of human diseases, including cancer, diabetes, and certain neurological disorders, methylation patterns have the potential for use as disease biomarkers.
What is Copy Number Variation (CNV)?
CNV refers to changes in the DNA copy number level of a region when compared to a reference genome. Recent research has shown that the level of CNV in the human genome is much higher than previously thought.
Other types of copy number changes occur when a region within a chromosome or an entire chromosome is amplified erratically or deleted altogether. Copy number aberrations are typical of cancer cells. In some tumors, high-level amplifications correlate with clinical outcome.
What is Loss of Heterozygosity (LOH)?
LOH represents the loss of a parent's contribution to part of the cell's genome. LOH is extremely common in cancer, where it often indicates the presence of a tumor suppressor gene in the region. Typically, the remaining copy of the tumor suppressor gene will be inactivated by a point mutation.
LOH can sometimes occur without apparent copy number changes (such as gene conversions), and recent publications have documented the critical role of copy-neutral LOH in tumor samples. Events such as these cannot be detected with traditional two-color microarray comparative genomic hybridization (array CGH).
Illumina DNA analysis solutions enable researchers to profile, at increased resolution, the genome for LOH, as well as other chromosomal aberrations routinely found in cancer and other disorders.
Genomics News
Geraldine Bliss wants parents to ask, “Could it be genetic?”
One genetic test changed everything for her son. Now Bliss, the founder of Start Genetic, wants more families to get answers sooner.
Read article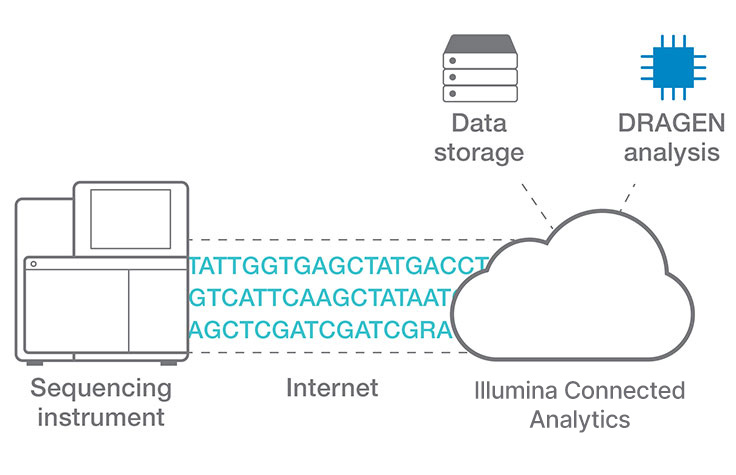
Efficient cloud data analysis for COPD multiomics project
Researchers at Okayama University in Japan use Illumina Connected Analytics with DRAGEN pipelines for analyzing whole-genome, exome, transcriptome, and metagenome data
Read Interview
Illumina gets behind the wheel to drive greater results
Across China, Illumina is delivering end-to-end research analysis support, scalable high-performance computing, and localized deployment
Read articleCan Applying NGS Lead to Better Beer?
An international group of scientists, yeast producers, and brewers sequenced 96 yeast strains to learn how genetics can influence the flavor of beer. Their findings, published in Cell, could help brewers make better beers.
Read Article