Paired-End vs. Single-Read Sequencing
What are the differences between paired-end and single-read sequencing?
Single-read sequencing involves sequencing DNA from only one end, and is the simplest way to utilize Illumina sequencing. Unlike single-read sequencing, paired-end sequencing allows users to sequence both ends of a fragment and generate high-quality, alignable sequence data. Paired-end sequencing facilitates detection of genomic rearrangements and repetitive sequence elements, as well as gene fusions and novel transcripts.
In addition to producing twice the number of reads for the same time and effort in library preparation, sequences aligned as read pairs enable more accurate read alignment and the ability to detect insertion-deletion (indel) variants, which is more difficult with single-read data.1 All Illumina next-generation sequencing (NGS) systems are capable of paired-end sequencing.
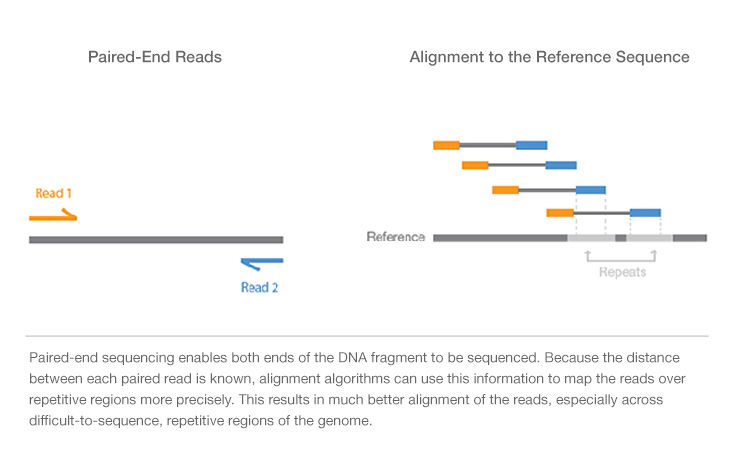
Paired-end vs. single-read sequencing advantages
Paired-end sequencing
- Simple paired-end libraries: Simple workflow allows generation of unique ranges of insert sizes
- Efficient sample use: Requires the same amount of DNA as single-read genomic DNA or cDNA sequencing
- Broad range of applications: Does not require methylation of DNA or restriction digestion; can be used for bisulfite sequencing
- Simple data analysis: Enables high-quality sequence assemblies with short-insert libraries. A simple modification to the standard single-read library preparation process facilitates reading both the forward and reverse template strands of each cluster during one paired-end read. Both reads contain long-range positional information, allowing for highly precise alignment of reads.
Single-read sequencing
- Cost-effective uses: This solution delivers large volumes of high-quality data, rapidly and economically
- Specific applications: Single-read sequencing can be a good choice for certain methods such as small RNA-Seq or chromatin immunoprecipitation sequencing (ChIP-Seq)
Genomic and transcriptomic paired-end sequencing
Paired-end DNA sequencing
Paired-end DNA sequencing reads provide high-quality alignment across DNA regions containing repetitive sequences, and produce long contigs for de novo sequencing by filling gaps in the consensus sequence. Paired-end DNA sequencing also detects common DNA rearrangements such as insertions, deletions, and inversions.
Paired-end RNA sequencing
Paired-end RNA sequencing (RNA-Seq) enables discovery applications such as detecting gene fusions in cancer and characterizing novel splice isoforms.2
For paired-end RNA-Seq, Illumina offers kits with an alternate fragmentation protocol, followed by standard Illumina paired-end cluster generation and sequencing.
Related methods
DNA sequencing
DNA sequencing can be applied to genes, DNA regions of interest, or the entire genome through a variety of methods.
RNA sequencing
RNA-Seq offers a high-resolution view of coding and noncoding regions of the transcriptome for a deeper understanding of biology.

High-impact discovery through gene expression and regulation research
Explore how RNA sequencing technologies, along with complementary techniques such as single-cell and spatial RNA-Seq, protein, chromatin, and methylation analysis, are impacting our understanding of biology and disease.
Related content
Next-generation sequencing technology
Discover the broad range of experiments you can perform with NGS, and explore major sequencing technology advances.
ChIP assays with NGS sequencing
Learn how you can get unbiased, genome-wide insights into gene regulation using ChIP-Seq.
Whole-exome sequencing
Focus on studying the protein-coding regions of the genome to detect exonic variants and uncover genetic influences on disease and population health.
mRNA sequencing
Get a comprehensive view of the coding transcriptome. Detect both known and novel transcripts and measure transcript abundance.
Sequencing read length
Choosing the right sequencing read length depends on your sample type, application, and coverage requirements. Learn how to calculate the right read length for your sequencing run.
Educational webinars
Applications and advances in whole-genome sequencing
Learn about the latest advances in whole-genome sequencing and the vast potential of emerging genomics technologies.
Redefining NGS in cancer research
See how cancer researchers are using NGS to characterize the proteome, epigenome, non-coding RNAs, and even small RNAs.
From sample to genomic insights
Mile Lelivelt, VP of Product Management for Software and Informatics, shares his thoughts on Illumina solutions to arrive at critical genomic insights.
Featured products
Illumina Stranded mRNA Prep
A simple, scalable, cost-effective, rapid single-day solution for analyzing the coding transcriptome.
Illumina Stranded Total RNA Prep with Ribo-Zero Plus
Rapid library preparation from a broad range of sample types for studying the coding and non-coding transcriptome with exceptional study flexibility.
Interested in receiving newsletters, case studies, and information from Illumina based on your area of interest? Sign up now.
Additional resources
Illumina sequencing by synthesis in action
See how Illumina sequencing by synthesis (SBS) chemistry works.

Illumina sequencing platforms
View benchtop and production-scale sequencers and find resources designed to help you choose the right platform.

Short paired-end vs. long single-end reads for expression analysis
Learn about the advantages of short paired-end reads over long single-end reads for RNA-Seq in this paper.

Library preparation
Innovative, comprehensive library prep solutions are a key part of the Illumina sequencing workflow.
References
- Nakazato T, Ohta T, Bono H. Experimental design-based functional mining and characterization of high-throughput sequencing data in the sequence read archive. PLoS One. 2013;8(10):e77910.
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63.